|  |
|  |
|  |
| |
| |
|  |
|


Welcome
to the Computer Vision Research Laboratory at UCSB. The name is there for
historical reasons; however, our research agenda and project scope have
broadened to include a diverse set of interesting and challenging topics.
Today, students, visitors, and faculty are engaged in advanced research related
to computer vision, medical image analysis, computer graphics, and
bioinformatics. This page is constantly under construction and contains
descriptions of some sample projects in the Computer Vision Laboratory. If you
desire further information, please contact Professor Yuan-Fang Wang directly.

Modeling the 3D Scenes using a single COTS
(commercial-off-the-shelf) digital camera or camcorder

Using a digital camera or camcorder, one can take multiple
pictures at random locations in a 3D scene or around some objects of interest.
The goal is to model the appearance, structure and behavior (e.g., deformation
and motion) of the 3D scenes/objects.
There are, in fact, a multitude of possible formulations of the
3D modeling problem. On one extreme, the input images can be stitched together
to build a panorama without explicitly inferring the 3D depth. On the other
extreme, 3D depth and texture can be densely recovered at each and every pixel
location. Certainly, middle-of-the-road solutions, such as inferring only the
camera motion but not the object structure, or inferring discrete, sparse 3D
structures, are also possible.
Our research proposes a spectrum of such solutions, from image
stitching, to spatially-aware image browsing, to sparse 3D structures
("point cloud" representation), to dense textured-surface 3D models.
Our techniques work with any commercial-off-the-shelf digital cameras, without
elaborate calibration, special photography equipment, or active sensors. Our
modeling algorithms automatically analyze the images to deduce the 3D structure
and appearance of the scene/objects. Many 360-degree (complete) and partial 3D
models can be constructed this way. Please see our demo pages for more information.
If you desire to
try your own photos with our programs, please go here.
(A new browser page will open to take you to a site off the UCSB domain.)
![]()
![]()
 A Video Analysis Framework for
Soft Biometry Security Surveillance
A Video Analysis Framework for
Soft Biometry Security Surveillance
We propose a distributed, multi-camera video
analysis paradigm for airport security surveillance. We propose to use a new
class of biometry signatures, which are called soft biometry including a
person's height, built, skin tone, color of shirts and trousers, motion
pattern, trajectory history, etc., to ID and track errant passengers and
suspicious events without having to shut down a whole terminal building and
cancel multiple flights. One might suspect, and we concur, that it is not very
difficult to compute some of these soft biometric signatures from individual,
properly-segmented image frames. The real challenge, however, is in designing a
robust and intelligent video-analysis system to support the reliable acquisition,
maintenance, and correspondence of soft biometry signatures in a coordinated
manner from a large number of video streams gathered in a large camera network.
The intellectual merit of the proposed research is to address three important
video analysis problems in a distributed, multi-camera surveillance network:
sensor network calibration, peer-to-peer sensor data fusion, and
stationary-dynamic cooperative camera sensing.
q Sensor network
calibration. In order to correctly correlate and fuse
information from multiple cameras, calibration is of paramount importance.
Cameras deployed in a large network have different physical characteristics,
such as location, field-of-view (FOV), spatial resolution, color sensitivity,
and notion of time. The difference makes answering even simple queries
exceedingly difficult. For example, if a subject moves from the FOV of one
camera to another, which has different color sensitivity and operates under
dissimilar lighting conditions, drastic changes in color signatures do occur.
To reliably compute soft biometry to assist the identification of subjects
across the FOVs of multiple cameras therefore
requires careful color calibration. We have developed and integrated a suite of
algorithms for spatial, temporal, and color calibration for cameras with both
overlapped and non-overlapped FOVs.
q Peer-to-peer sensor
data fusion. As cameras have limited FOVs,
multiple cameras are often stationed to monitor an extended surveillance area,
such as an indoor arrival/departure lounge or an outdoor parking lot.
Collectively, these cameras provide complete spatial coverage of the
surveillance area. (A small amount of occlusion by architectural fixtures,
decoration, and plantation is often unavoidable.) Individually, the event
description inferred from a single camera is likely to be incomplete. (E.g.,
the trajectory of a vehicle entering a parking lot is only partially observed
from a certain vantage point.) We have developed algorithms to fuse video data
from multiple cameras for reliable event detection using a hierarchy of Kalman Filters.
q Stationary-dynamic
cooperative camera sensing. To achieve
effective wide-area surveillance with limited hardware, a surveillance camera
is often configured to have a large FOV. However, once suspicious persons/activities
have been identified through video analysis, selected cameras ought to obtain
close-up views of these suspicious subjects for further scrutiny and
identification (e.g., to obtain a close-up view of the license plate of a car
or the face of a person). Our solution is to employ stationary-dynamic camera
assemblies to enable wide-area coverage and selective focus-of-attention
through cooperative sensing. That is, the stationary cameras perform a global,
wide FOV analysis of the motion patterns in the surveillance zone. Based on
some pre-specified criteria, the stationary cameras identify suspicious
behaviors or subjects that need further attention. The dynamic camera, mounted
on a mobile platform and equipped with a zoom lens, is then used to obtain
close-up view of the subject to reliably compute soft biometry signatures. We
have studied research issues to enable cooperative camera sensing, including
dynamic camera calibration and stationary-dynamic camera sensing using a visual
feedback paradigm.
![]()
![]()
 Toward
Automated Reconstruction of 2D and 3D Scenes from Video Images
Toward
Automated Reconstruction of 2D and 3D Scenes from Video Images
In this project, we
study the problem of automated reconstruction of 2D and 3D scene (structure,
appearance, and behavior) from video images. While there are many similar
projects being conducted at academia and industry, our project addresses a
number of difficult issues in (1) the 3D structures can vary significantly from
almost planar to highly complex with large variation in depth, (2) the camera
can be at varying distances to the scene, and (3) the scene may show
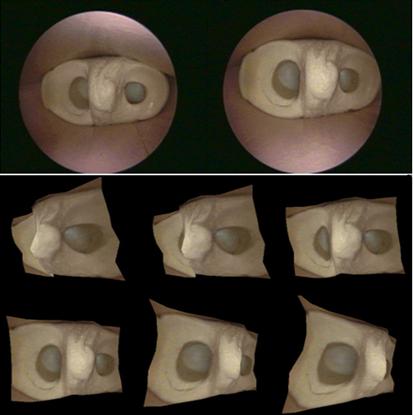
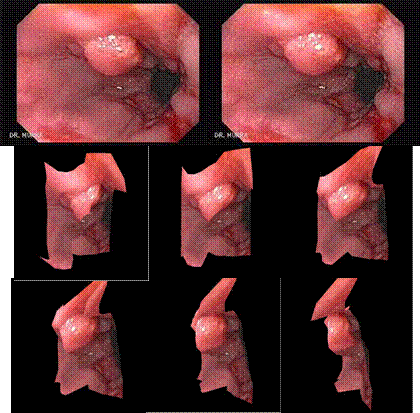
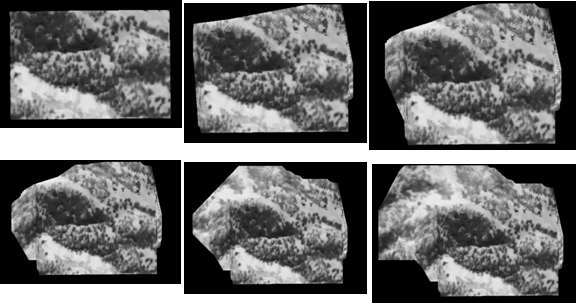
significant deformation over time. Some sample results are shown below. In the
first two examples, two
images (the top row) were used to generate a 3D model of the visible surface
structure with correct texture mapping. Novel views can then be synthesized
from the reconstructed 3D model (the bottom rows). These images are taken from
inside a knee mockup and from real endoscopy surgery. The fourth example shows
the stitching of terrain model from UAV (unmanned aerial vehicle) flight data.
![]()
![]()
Robust and Real-Time Image Stabilization and Rectification
We
study a unified framework for achieving robust and real-time image
stabilization and rectification. While compensating for a small amount of image
jitter due to platform vibration and hand tremble is not a very difficult task,
canceling a large amount of image jitter, due to significant, long-range, and
purposeful camera motion (such as panning, zooming, and rotation), is much more
challenging. While the terms “significant,
long-range, and purposeful” may
imply that we should not cancel this motion, it should be remembered that in
many real-world imaging systems there may be multiple objectives with
conflicting solutions. By this we mean that while significant and purposeful
camera manipulation is needed to explore new perspectives and acquire novel
views, such manipulation often times causes difficulty for the human operator
in image interpretation. Hence, an image stabilization algorithm should be
designed to allow significant freedom in image acquisition while alleviating difficulty in image interpretation. We mention here two practical problems in
diverse application domains that can make use of such an image stabilization
and rectification algorithm.
q
The
first application is in rectifying the video display in video-endoscopy.
Endoscopes procedures are minimally invasive surgical procedures where several
small incisions are made on the patient to accommodate surgical instruments
such as scalpels, scissors, staple guns, and an endoscope. The scope acquires
images of the bodily cavity that are displayed in real time on a monitor to
provide the visual feedback to the surgeon to perform surgery. In order to view
the anatomy in a highly constrictive body cavity (e.g., nasal passage in rhinoscopy and inner ear cavity in otoscopy)
and subject to the entry point constraint, the surgeon often manipulates the
scope with large panning and rotation motion to eliminate blind spots. The
views acquired can be highly non-intuitive, e.g., the anatomy can appear with
large perspective distortion, sideways, or even upside down. Hence, while this
type of manipulation is necessary to reveal anatomical details, it does cause
significant difficulty in image interpretation.
q The second application is in
rectifying the video display in an unmanned aerial vehicle (UAV). Under
the control of a ground operator, an UAV may purposefully pitch, roll, and
rotate to maneuver into certain positions or to evade ground fire. Executing
such maneuvers severely alters the capture angle of the on-board camera. Again,
the banking action is purposefully aiding in the flight but hindering the
intuitive nature of the viewed video.
As
should become clear from the preceding discussion, in both applications, the
operator manipulates the camera using large panning, zooming, and rotating
actions to obtain better views of the subjects (i.e., organs and ground
vehicles). The views thus displayed can be highly non-intuitive, may have large
perspective or other types of distortion, and may even be upside down. The
freedom in such manipulation is absolutely necessary and should not be
restricted solely for easing the difficulty in image interpretation. Instead,
the goal in designing image rectification algorithms for such applications
should be to maintain some consistency and uniformity in the display, while allowing
the operator to survey the scene as before.
Our framework selectively compensates for
unwanted camera motion to maintain a stable view of the scene. The rectified
display has the same information content, but is shown in a much more
operator-friendly way. Our contribution is
three-fold: (1) proposing a unified image rectification algorithm to cancel
large and purposeful image motion to achieve a stable display that is
applicable for both far-field and near-field image conditions, (2) improving
the robustness and real-time performance of these algorithms with extensive
validation on real images, and (3) illustrating the potential of these
algorithms by applying them to real-world problems in diverse application
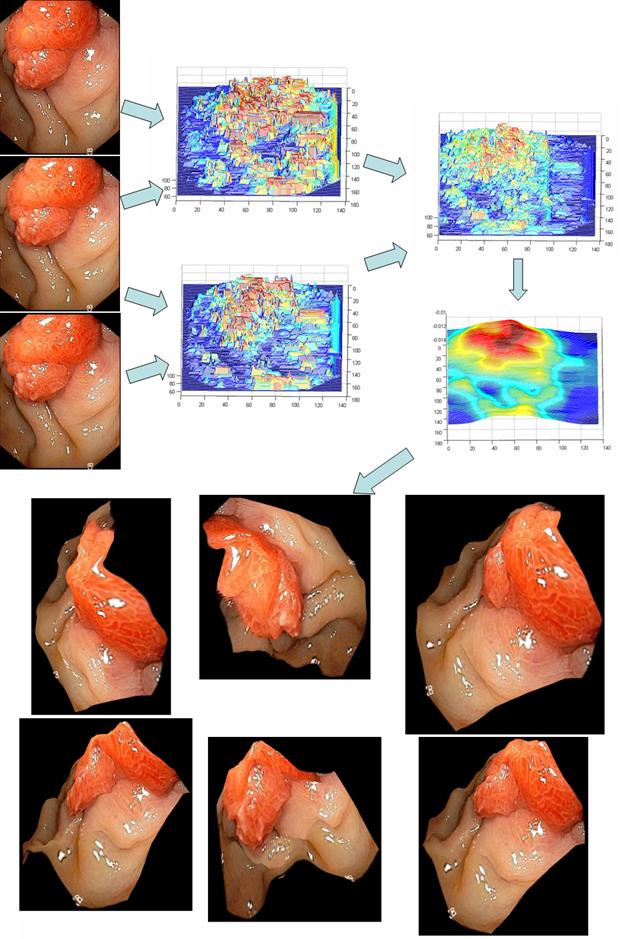
domains. The
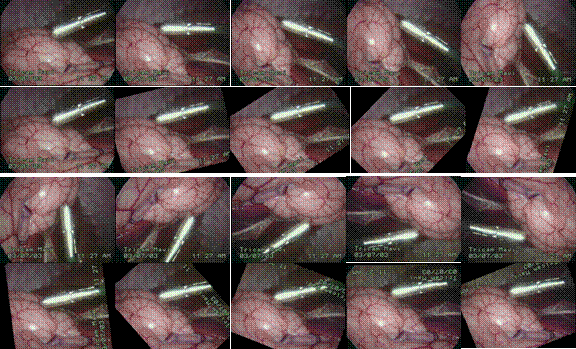
following figures show some sampling results (from real endoscopic surgery and
UAV flight data). Video sequences are shown from left to right and from top to
bottom. The display is grouped by showing the original, uncertified images on
top and the corresponding rectified images immediately on the bottom. As can be
seen that even with large panning, zooming and rotation (1st and 3rd
rows), our algorithm is able to maintain the orientation in the rectified
images (2nd and 4th rows).
![]()
![]()
Toward Real-time, Physically-correct Soft Tissue Behavior
Simulation
We present a behavior simulation algorithm
that has the potential of enabling physically-correct, photo-realistic, and
real-time behavior simulation for soft tissues and organs. Our approach
combines a physically-correct formulation based on boundary element methods
with an efficient numeric solver. There are many scenarios, both in off-line
training of surgeons and on-line computer-assisted surgery, that the proposed
technique can be useful. For example, simulators, used for the purpose of
training surgeons in the pre-operative stage, require physically-correct,
real-time response of the graphical rendering to inputs from the trainee.
However, it has long been recognized that it is difficult to simultaneously
satisfy the requirements of physical-correctness and real-time. It is well
known that simulation of deformable behaviors is difficult. So far, approaches
to this problem can be roughly classified into two categories: those that aim
more at efficiency and those that aim more at accuracy. The former is
categorized by many mass-spring, spline, and superquadric
models while the latter mainly comprises techniques based on the finite element
methods (FEM). Our modeling scheme aims to achieve the best of both worlds by
providing necessary accuracy at a speed comparable to that of the efficient
models.
Our method is based on a physically corrected
formulation based on boundary element methods (BEM). Naively speaking, BEM for
structure simulation concentrates the analysis power on the boundary of the
object (or the surface of a 3D organ). Intuitively, for the same level of
discrete resolution, the BEM-based methods have the potential of being significantly
less expensive than their FEM-based counterparts. This is because that the FEM
methods employ O(n^3) variables (representing the
displacement of the body at a particular point under externally applied forces
and torques), scattered both in the interior and on the boundary of the object.
The BEM methods employ O(n^2) variables (representing
surface displacement and traction) only on the boundary of the object, with n representing the resolution along a
particular dimension. Hence, BEM achieves significant saving in terms of
problem size. (One might argue that there are ways to reduce the problem size
for FEM, e.g., multi-resolution and adaptive grid. We are aware of the
possibilities. The above analysis serves only as an illustration and does not
mean to ignore these possibilities. Furthermore, similar efficient numeric
techniques are often applicable to BEM to achieve a corresponding reduction in
problem size as well.)
While this simple analysis might look
promising, the reality is never this straightforward. No matter it is an FEM-
or a BEM-based simulation, the gist of the simulation all comes down to solving
a system of linear equations of the form AX=B
over time (or a time marching problem), where A involves the material properties such as the Lame constants (and
other quantities that have to do with the discretization
and interpolation functions in the elements), B involves known boundary conditions (e.g., known displacement and
traction at certain surface and interior points), and X are the unknown displacement and traction inside and on the
surface of the object. Numeric analysts will quickly point out that while BEM
requires less number of variables – which results in a much smaller system of
equations (O(n^2) for BEM vs. O(n^3) for FEM) – the real complexity of the BEM
solution can be higher. This is because that while FEM results in a bigger
matrix A, A is often well conditioned and sparse. Efficient solutions exist
for many classes of well conditioned, sparse matrices, which bring the
complexity of solution to O(n^3). BEM, on the other
hand, always results in a dense matrix A,
and the complexity of the solution can be proportional to the cube of the
matrix size.
Our proposed numeric algorithm exploits a mechanism
to reduce matrix update and solve complexity. Our observation is that the
kernel function in the fundamental solution of the BEM is usually smooth, which
results in the coefficient matrix having a block-wise low-rank structure, which
we call sequentially semi-separable (SSS) for the one-dimensional case, and
hierarchically semi-separable (HSS) for higher dimensions. Exploiting this
particular matrix structure in our simulation, we are able to achieve real-time
behavior simulation on ordinary PC of fairly complex organs. Our simulation
allows large changes in boundary conditions, such as those resulted from
organ-organ and organ-body wall collision, which is not possible in current
state-of-the-art using BEM. This is a significant improvement that increases
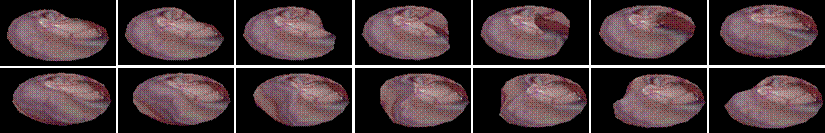
the applicability of the BEM methods. Snap shots of two deformation sequences
are shown below.
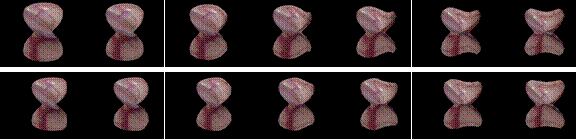
 Sample
video clips (These files are in Windows Media Player wmv format. To save download bandwidth, the video clips are
fairly short. They demonstrate an organ being deformed by spatially- and
temporally-varying large disturbance with the volume preserved.)
Sample
video clips (These files are in Windows Media Player wmv format. To save download bandwidth, the video clips are
fairly short. They demonstrate an organ being deformed by spatially- and
temporally-varying large disturbance with the volume preserved.)
A spherical organ under a poking disturbance. (584k)
The same spherical organ under a rubbing disturbance. (560k)
The
same sphere organ under a combined rubbing and poking disturbance. (631k)
Extremely large deformation.
(2.5M)
![]()
![]()
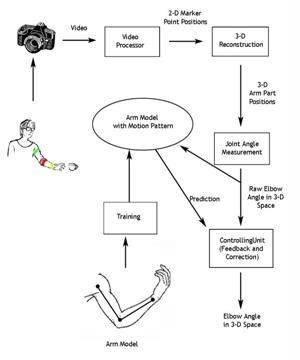
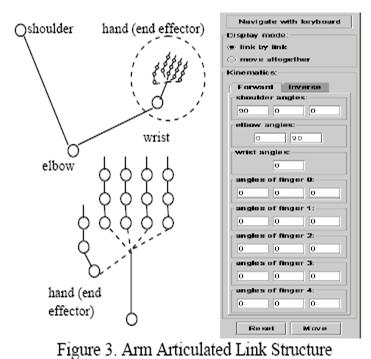
A System for Limb Modeling, Position Sensing and Stimulation
Control
We are
investigating a system for sensing, modeling and control of an upper extremity
neural prosthesis. The sensing unit employs a computer vision approach wherein
one or more video cameras are used to detect movement of the arm and provide
the arm position information to a model. The model uses kinematics and dynamics
simulation to control the stimulation and animation of the articulated links.
The motion control unit integrates a priori knowledge from the trained model
and the observed sensor input, smoothes the limb motion tracking results and
delivers a feedback signal to guide or correct the sensing process. In our
experiments we compared sensed elbow angle accuracy results between our
computer vision based system and developed a
visualization system for the arm model.
![]()
![]()
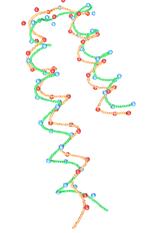
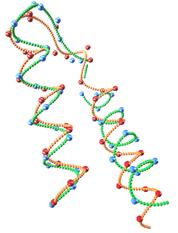
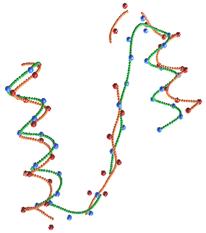
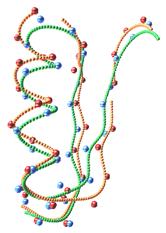
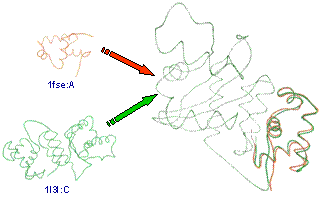 Protein Structure Alignment and Fast Similarity Search Using Local
Shape Signatures
Protein Structure Alignment and Fast Similarity Search Using Local
Shape Signatures
The number of known protein structures is increasing
rapidly, as more researchers are joining the hunt for novel protein structures,
more experimental apparatus are deployed, and more theoretical frameworks and
software tools are developed for predicting protein structures. Protein
structure comparison tools play an important role in this enterprise. In
predicting a protein structure from its sequence, researchers usually form a
new candidate structure. To avoid potential exponential explosion of
structures, that new structure is compared with previously known structures for
verification/tuning/correction. Discovering similar folds or similar
substructures thus provides restrictions on the conformational space and serves
as a starting point for producing useful models.
We present a new method for conducting protein
structure similarity searches, which improves on the accuracy, robustness, and
efficiency of some existing techniques. Our method is grounded in the theory of
differential geometry on 3D space curve matching. We generate shape signatures
for proteins that are invariant, localized, robust, compact, and biologically
meaningful. To improve matching accuracy, we smooth the noisy raw atomic
coordinate data with spline fitting. To improve matching efficiency, we adopt a
hierarchical coarse-to-fine strategy. We use an efficient hashing-based
technique to screen out unlikely candidates and perform detailed pairwise alignments only for a small number of candidates
that survive the screening process. Contrary to other hashing based techniques,
our technique employs domain specific information (not just geometric
information) in constructing the hash key, and hence, is more tuned to the
domain of biology. Furthermore, the invariancy,
localization, and compactness of the shape signatures allow us to utilize a
well-known local sequence alignment algorithm for aligning two protein
structures. One measure of the efficacy of the proposed technique is that we
were able to perform structure alignment queries 30 times faster than a
well-known method while keeping the quality of the query results at the same
level.
![]()
![]()
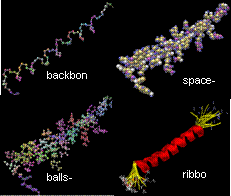
 FPV: Fast
Protein Visualization Using Java3D
FPV: Fast
Protein Visualization Using Java3D
The ability to visualize the 3D structure of
proteins is critical in many areas such as drug design and protein modeling.
This is because the 3D structure of a protein determines its interaction with
other molecules, hence its function, and the relation of the protein to other
known proteins. We have developed a protein visualization system based on Java
3D. There is growing trend in adopting the Java technology in the fields of
bioinformatics and computational biology. The main advantages of Java are its
compatibility across different systems/platforms and having the ability to be
run remotely through web browsers. Using Java 3D as a graphics engine has also
the additional advantage of rapid application development, because Java 3D API
incorporates a high-level scene graph model that allows developers to focus on
the objects and the scene composition. Java 3D also promises high performance,
because it is capable of taking advantage of the graphics hardware in a system.
However, using Java 3D for visualization has some performance issues with it.
The primary concerns about molecular visualization tools based on Java 3D are
in their being slow in terms of interaction speed and in their inability to
load large molecules. This behavior is especially apparent when the number of
atoms to be displayed is huge, or when several proteins are to be displayed
simultaneously for comparison. In this project we present techniques for
organizing a Java 3D scene graph to tackle these problems. We demonstrate the
effectiveness of these techniques by comparing the visualization component of
our system with two other Java 3D based molecular visualization tools. In
particular, for van der Waals display mode, with the
efficient organization of the scene graph, we could achieve up to eight times
improvement in rendering speed and could load molecules three times as large as
the previous systems could.
![]()
![]()

 Efficient
Molecular Surface Generation Using Level-Set Methods
Efficient
Molecular Surface Generation Using Level-Set Methods
Molecules interact through their surface
residues. Calculation of the molecular surface of a protein structure is thus
an important step for a detailed functional analysis. One of the main
considerations in comparing existing methods for molecular surface computations
is their speed. Most of the methods that produce satisfying results for small
molecules fail to do so for large complexes. In this project we present a
level-set-based approach to compute and visualize a molecular surface at a
desired resolution. The emerging level-set methods have been used for computing
evolving boundaries in several application areas from fluid mechanics to
computer vision. We use a level-set-based approach to compute the molecular
surface of a protein of known structure. Our method proceeds in three
stages:(1) An outward propagation step that generates the van der Waals surface and the solvent-accessible surface, (2) An
inward propagation step that generates the re-entrant surfaces and contact
surfaces, i.e., the solvent excluded or the molecular surface, and (3) Another
inward propagation step to determine the outer surface and interior cavities of
the molecule. The novelty of our algorithm is three-fold: First, we propose a
unified framework for solving all the tasks above based on the level-set
front-propagation method; second, our algorithm traverses each grid cell at most once and never visits grid cells
that are outside the sought-after surfaces to guarantee efficiency; and third,
our algorithm correctly detects interior cavities for all kinds of protein
topologies. Our method is able to calculate the surface and interior
inaccessible cavities very efficiently even for very large molecular complexes.
We compared our method to some of the most widely used molecular visualization
tools (Swiss-PDBViewer, PyMol,
and Chimera) and our results show that we can calculate and display a molecular
surface 1.5 to 3.14 times faster on average than all three of the compared
programs. Furthermore, we demonstrate that our method is able to detect all of
the interior inaccessible cavities that can accommodate one or more water
molecules.
![]()
![]()
 Automated Protein Classification Using Consensus Decision
Automated Protein Classification Using Consensus Decision
Protein classification is important as a protein’s
label often times gives a good indication to its biological function. Of many
existing classification schemes, SCOP is probably the most trusted one (as it
involves significant manual inspection). However, SCOP classification is labor
intensive and is not updated frequently. Hence, there is a desire to be able to
predict, through some automated means, the SCOP classification of new proteins.
A multitude of techniques, based on both sequence and structure similarity, can
be used for predicting SCOP classification. However, their applicability and
accuracy vary, depending both on the level of taxonomy (family, superfamily, or fold level) and the parameter settings of
the techniques. Hence, the classification results from multiple techniques
often show varying degrees of conformity with the manually-generated SCOP
classifications (the ground truth) and with one another.
This project is aimed at improving the
accuracy of automated SCOP classification by combining the decisions of
multiple methods in an intelligent manner, using the consensus of a committee
(or an ensemble) classifier. Our technique is rooted in machine learning that
shows that by judicially employing component classifiers, an ensemble
classifier can be constructed to outperform its components. We use two
sequence- and three structure-comparison tools as component classifiers. Given
a protein structure, using the joint hypothesis we first determine if the
protein belongs to an existing group (family, superfamily,
or fold) in the SCOP hierarchy. For the proteins that are predicted as members
of the existing groups, we then compute their family-, superfamily-,
and fold-level classifications using the consensus classifier.
We have shown that by using this method we
can significantly improve the classification accuracy compared to those of the
individual component classifiers. In particular, we have achieved error rates
that are 3 to 12 times less than the individual classifiers' error rates at the
family level, 1.5 to 4.5 times less at the superfamily
level, and 1.1 to 2.4 times less at the fold level. Our method achieves 98%
success for family assignments, 87% success for superfamily
assignments, and 61% success for fold assignments. What is significant is that
these accuracy numbers are very close to the theoretically maximum performance
achievable through ensemble combination.
![]()
![]()
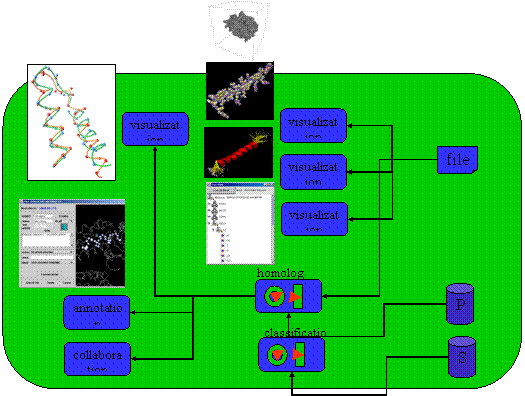
 Personalized Annotation and Information Sharing in Protein Science
Personalized Annotation and Information Sharing in Protein Science
In this project, we develop a software tool called Information-slips (abbr. as i-slips) that provides a convenient and
customizable mechanism for remote collaboration and data sharing in protein
science. I-slips are small 3D objects that coexist with and
augment the host 3D objects (in our application, protein models). Each
i-slip keeps track of three types of information: (1)
the host object, which is part (e.g., an amino acid or a secondary structure
element) of a 3D protein, (2) the visual configuration, which defines the way
to visualize the i-slip, and (3) the content object,
which stores user annotation and augmentative information about the host
object. Our i-slip design
makes two main contributions. Firstly, i-slip goes
beyond simple passive annotation to also provide active interactivity. The
content object can embed an action to perform a user-defined operation
on-demand. Furthermore, the condition to perform the embedded action can be
event-driven. That is, i-slips can monitor the
environment and automatically handle such predefined events when they occur
without the user intervention. Secondly, i-slip is a
highly versatile and adaptable information container. The user can easily
customize i-slip templates to support new
domain-specific content objects, and develop new actions to embed
domain-dependent algorithms. Furthermore, the storage and transportation of i-slips are based on the XML technology for a high degree
of interoperability. To the best of our knowledge, this is the first protein
visualization tool that supports user-contributed information and embeddable actions/activities.
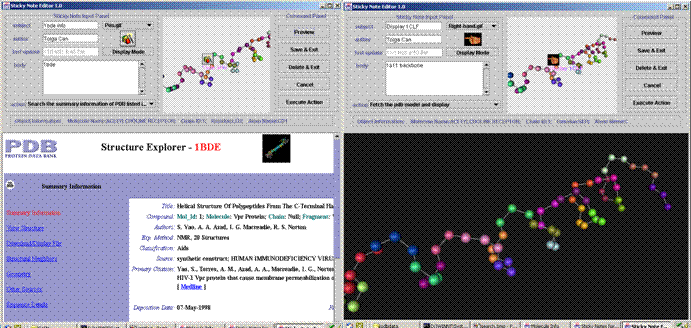
The following figures show our system in action. The left one shows that an
action note can automatically query PDB database to download and display the
PDB file of the protein. The right figure shows a structure comparison action
note that can retrieve from PDB and display proteins of a similar structure as
the one being studied.
![]()
![]()


 3D Model Construction by Fusing Heterogeneous Sensor
Data
3D Model Construction by Fusing Heterogeneous Sensor
Data
In
this project, we propose a scheme for 3D model construction by fusing
heterogeneous sensor data. The proposed scheme is intended for use in an
environment where multiple, heterogeneous sensors operate asynchronously.
Surface depth, orientation, and curvature measurements obtained from multiple
sensors and vantage points are incorporated to construct a computer description
of the imaged object. The proposed scheme uses Kalman
filter as the sensor data integration tool and hierarchical spline surface as
the recording data structure. Kalman filter is used
to obtain statistically optimal estimates of the imaged surface structure based
on possibly noisy sensor measurements. Hierarchical spline surface is used as
the representation scheme because it maintains high-order surface derivative
continuity, may be adaptively refined, and is storage efficient. We show in
this project how these mathematical tools can be used in designing a modeling
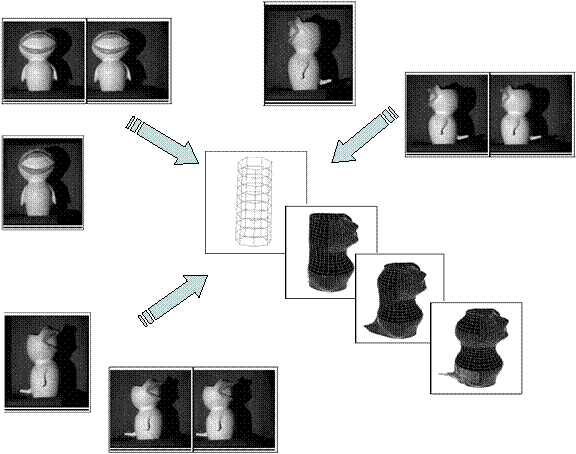
scheme to fuse heterogeneous sensor data. A sample result is included below,
where two types of sensor data (video and structured-light coded) from three
vantage points were gathered sequentially. 3D surface
structures were then iteratively constructed and refined by incorporating these
sensor data.
 Automated
Instrument Tracking in Robotically-Assisted Laparoscopic Surgery
Automated
Instrument Tracking in Robotically-Assisted Laparoscopic Surgery
This
project designs and implements a practical and reliable image analysis and tracking
algorithm to achieve automated instrument localization and scope maneuvering in
robotically-assisted laparoscopic surgery. Laparoscopy is a minimally invasive
surgical procedure which utilizes multiple small incisions on the patient's
body through which the surgeon inserts tools and a video scope for conducting
an operation. The scope relays images of internal organs to a camera and the
images are displayed on a video screen. The surgeon performs the operation by
viewing the scope images, as opposed to the traditional “open” procedure where
a large incision is made on the patient's body for direct viewing.
The
current mode of the laparoscopy has an assistant holding the scope and
positioning it in response to the surgeon's verbal commands. However, this
results in suboptimal visual feedback because the scope is often aimed
incorrectly and vibrates due to hand trembling. Computer Motion Inc. has
developed a robotic laparoscope positioner (see
picture above) to replace the assistant. The surgeon commands the robotic positioner through a hand/foot controller interface or a
voice recognition interface.
To
further simplify the man-machine interface in controlling the robotic scope positioner, we report here a novel scope positioning scheme
using automated image analysis and robotic visual servoing.
The scheme enables the surgeon to control his visual feedback and be more
efficient in performing surgery without
requiring additional use of the hands. For example, our technique
is able to automatically center and track instruments without the surgeon’s
voice command. This is important as for the safety of the patient, the surgeon
must see the operating instrument at all times. Our technique performs
automated image analysis to locate and track instruments in the image. When the
position of the instrument deviates from the desired location, (e.g., the
center of the image) or becomes too large or too small, our system
automatically generates the robot control signal to maneuver the robot to
obtain better viewing for the surgeon. The tracking system was incorporated
into the product offering of Computer Motion Inc. and was used in over
thousands of real-world surgery (Computer Motion Inc. has since merged with
Intuitive Surgical).


![]()
![]()

 Local Scale
Controlled Anisotropic Diffusion with Local Noise Estimate for Image Smoothing
and Edge Detection
Local Scale
Controlled Anisotropic Diffusion with Local Noise Estimate for Image Smoothing
and Edge Detection
This
project studies a novel local scale controlled piecewise linear diffusion for
selective smoothing and edge detection. The diffusion stops at the place and
time determined by the minimum reliable local scale and a spatial variant,
anisotropic local noise estimate. It shows nisotropic,
nonlinear diffusion equation using diffusion coefficients/tensors that
continuously depend on the gradient is not necessary to achieve sharp,
undistorted, stable edge detection across many scales. The new diffusion is
anisotropic and asymmetric only at places it needs to be, i.e., at significant
edges. It not only does not diffuse across significant edges, but also enhances
edges. It advances geometry-driven diffusion because it is a piecewise linear
model rather than a full nonlinear model, thus it is simple to implement and
analyze, and avoids the difficulties and problems associated with nonlinear
diffusion. It advances local scale control by introducing spatial variant,
anisotropic local noise estimation, and local stopping of diffusion. The
original local scale control was based on the unrealistic assumption of
uniformly distributed noise independent of the image signal. The local noise
estimate significantly improves local scale control.
![]()
![]()
ProteinVista
Many software programs have been developed to
visualize protein and molecular structures. However, as most visualization
software programs do not fully utilize 3D graphics hardware in personal
computers, visualization speed and rendering quality can be unsatisfactory. The
functionality provided by the script languages of these software programs might
also be incomplete to handle huge protein. We have developed a protein
visualization platform named ProteinVista. ProteinVista is a well-designed visualization system using
the Microsoft Direct3D graphics library and the Microsoft .net framework. It
provides a large variety of visualization styles and numerous visualization
options. It is also optimized for advanced 3D graphics hardware. The script
language used in Protein Vista is based on .net framework and can be written in
high-level languages such as C#. It provides powerful functionalities and is
easy to understand.
![]()
![]()
